Snakemake 是基于 Python 的一款工具,所以它也继承了 Python 语言简单易读、逻辑清晰、便于维护的特点,同时它还支持 Python 语法,非常适合新手用户。例如遵循python中缩进表示层级;以及索引从0开始,{input[0]}表示input里的第1个元素;列表用中括号类似[‘A’,’B’,’C’]等。snakemake 的基本组成单位叫“规则”,即 rule;每个 rule 里面又有多个元素(input、output、shell等)。它的执行逻辑就是将各个 rule 利用 input/output 连接起来,形成一个完整的工作流。
一.数据准备 以RNAseq的数据为例
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 snakefile_rnaseq ├── genome │ ├── gencode.v19.annotation.gtf │ └── hg19.fa ├── index │ └── hg19 │ ├── genome.1.ht2 │ ├── genome.2.ht2 │ ├── genome.3.ht2 │ ├── genome.4.ht2 │ ├── genome.5.ht2 │ ├── genome.6.ht2 │ ├── genome.7.ht2 │ ├── genome.8.ht2 │ └── make_hg19.sh ├── SRR957677.1_1.fastq.gz ├── SRR957678.1_1.fastq.gz ├── SRR957679.1_1.fastq.gz └── SRR957680.1_1.fastq.gz
二.创建工作流文件snakefile 工作流文件 Snakefile 文件名并不是固定的,位置也不是固定的。当你的工作流文件就在当前目录下,且名称正好是 Snakefile 时,就可以像上面的示例一样,不用指定具体的工作流文件,snakemake 会自动调取当前路径的名为 Snakefile 的文件去执行。如果你要执行其他路径的 Snakefile 或者其他的文件名的工作流文件,可以使用 -s 参数。将工作流文件命名为 py 文件,这样你在写 Python 代码时会有语法高亮。
--snakefile, -s 指定Snakefile,否则是当前目录下的Snakefile--dryrun, -n 不真正执行,一般用来查看Snakefile是否有错snakemake --dag | dot -Tpdf > dag.pdf 可视化
1 2 3 4 5 # 创建 Snakefile $ touch rnaseqflow.py $ snakemake -n -s rnaseqflow.py Building DAG of jobs... Nothing to be done.
三.创建第一条rule 1 2 3 4 5 6 7 8 9 10 11 12 13 rule fastqc: # 定义第一条规则,命名为 fastqc input: # input: 指定输入 fq='SRR957677.1_1.fastq.gz' output: # output: 指定输出 'SRR957677.1_1_fastqc.zip' log:#指定log文件 'SRR957677.1_1.log' params: #指定参数 outdir='qc' shell: 'fastqc {input[fq]} -o {params[outdir]} 1>{log[0]} 2>&1' # 指定执行方式,这里有三种执行方式:shell、run、script。run执行python脚本,shell执行Bash脚本。还可以用script来执行外部脚本。比如 script:"scripts/script.py"或 script:"scripts/script.R"。 #{input[fq]} 也可以写成{input[0]},同理{params[outdir]}可写成{params[0]}
参数-n为dry run,即不实际运行;-p为打印命令;-np即为只打印全部命令而不运行,-s 指定文件,否则是当前目录下的Snakefile,查看命令是否正确:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 $ snakemake -np -s rnaseqflow.py Building DAG of jobs... Job counts: count jobs 1 fastqc 1 [Fri Oct 23 07:51:24 2020] rule fastqc: input: SRR957677.1_1.fastq.gz output: SRR957677.1_1_fastqc.zip log: SRR957677.1_1.log jobid: 0 fastqc SRR957677.1_1.fastq.gz -o qc 1>SRR957677.1_1.log 2>&1 #这里可以看出命令和我们平时写的一样 Job counts: count jobs 1 fastqc 1 This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
四.使用通配符匹配多个输入文件 上面的 fastqc规则仅仅比对了一个样本,可是实际项目中有几十上百的样本时,我们就不能这样直接写样本名来运行了。snakemake 允许使用通配符(wildcard)来批量运行命令。
rule all的input是流程最终的目标文件,类似于GNU Make,从顶部指定目标 。
expand是Snakemake的特有函数,类似列表推导式。
expand(‘{sample}.txt’,sample=SAMPLES)相当于[‘{sample}.txt’.format(sample=sample) for sample in SAMPLES]。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 (SAMPLES,)= glob_wildcards("{sample}.1_1.fastq.gz") # 这里也可运用列表一一列出来SAMPLES=['SRR957677' ,'SRR957678' ,'SRR957679' ,'SRR957680' ] rule all: input: expand('{sample_name}.1_1_fastqc.zip',sample_name=SAMPLES) rule fastqc: input: fq='{sample_name}.1_1.fastq.gz' output: '{sample_name}.1_1_fastqc.zip' log: '{sample_name}.1_1.log' params: outdir='qc' shell: 'fastqc {input[fq]} -o {params[outdir]} 1>{log[0]} 2>&1'
dry run 一下,可以加上 -p 参数让终端打印出 shell 运行的命令:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 $ snakemake -np -s rnaseqflow.py Building DAG of jobs... Job counts: count jobs 1 all 4 fastqc 5 [Fri Oct 23 08:04:34 2020] rule fastqc: input: SRR957677.1_1.fastq.gz output: SRR957677.1_1_fastqc.zip log: SRR957677.1_1.log jobid: 1 wildcards: sample_name=SRR957677 fastqc SRR957677.1_1.fastq.gz -o qc 1>SRR957677.1_1.log 2>&1 [Fri Oct 23 08:04:34 2020] rule fastqc: input: SRR957679.1_1.fastq.gz output: SRR957679.1_1_fastqc.zip log: SRR957679.1_1.log jobid: 3 wildcards: sample_name=SRR957679 fastqc SRR957679.1_1.fastq.gz -o qc 1>SRR957679.1_1.log 2>&1 [Fri Oct 23 08:04:34 2020] rule fastqc: input: SRR957680.1_1.fastq.gz output: SRR957680.1_1_fastqc.zip log: SRR957680.1_1.log jobid: 4 wildcards: sample_name=SRR957680 fastqc SRR957680.1_1.fastq.gz -o qc 1>SRR957680.1_1.log 2>&1 [Fri Oct 23 08:04:34 2020] rule fastqc: input: SRR957678.1_1.fastq.gz output: SRR957678.1_1_fastqc.zip log: SRR957678.1_1.log jobid: 2 wildcards: sample_name=SRR957678 fastqc SRR957678.1_1.fastq.gz -o qc 1>SRR957678.1_1.log 2>&1 [Fri Oct 23 08:04:34 2020] localrule all: input: SRR957677.1_1_fastqc.zip, SRR957678.1_1_fastqc.zip, SRR957679.1_1_fastqc.zip, SRR957680.1_1_fastqc.zip jobid: 0 Job counts: count jobs 1 all 4 fastqc 5 This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
本文的例子都是各个样品批量、并行运行同样的步骤,所以可以全部使用通配符 {sample} 完成匹配;但有的步骤需要将多个样本的bam文件传递给一个命令而不再并行,这种方法就不在适用了。例如在以变异检测为例。
1. 首先在工作流文件开头定义一个变量:
1 samples = ["A", "B", "C"]
2. 然后使用 snakemake 的内置函数 expand:
1 bam=expand("mapped/{sample}.sorted.bam", sample=samples)
1 2 3 4 5 6 7 8 9 10 11 rule calling: input: fa="data/genome.fa", bam=expand("mapped/{sample}.sorted.bam", sample=samples), bai=expand("mapped/{sample}.sorted.bam.bai", sample=samples) output: "calling/all.vcf" shell: "samtools mpileup -g -f {input.fa} {input.bam} | " "bcftools call -mv - > {output}"
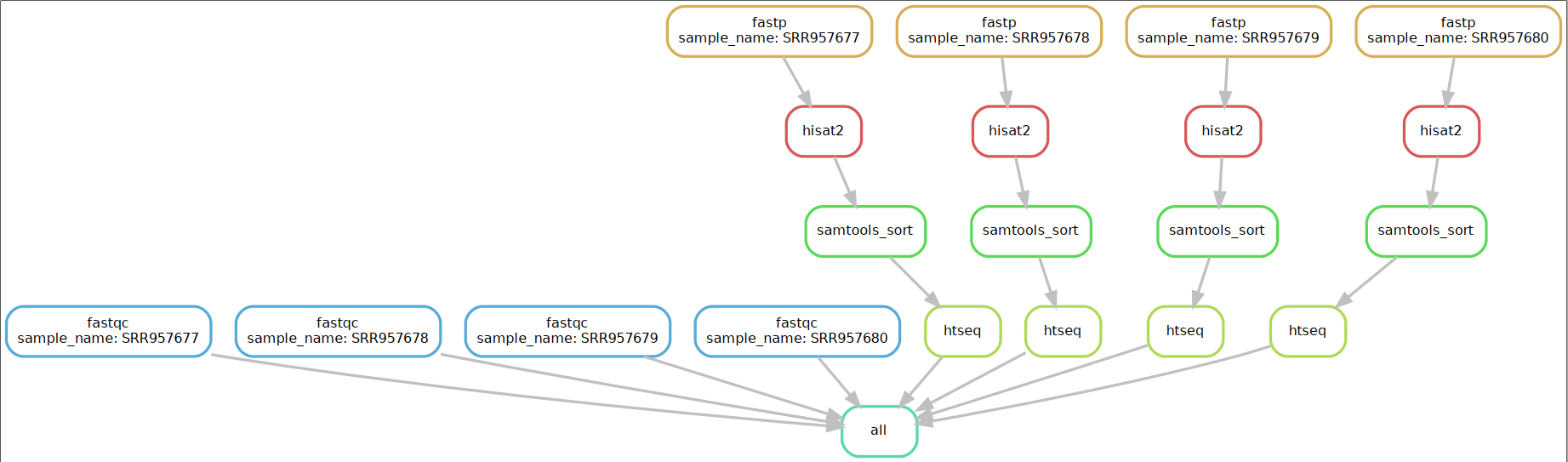
五.添加其他rule 在fastqc的基础上添加fastp、hisat2、samtools sort、htseq等rule。前后相连的rule,前一个rule的输出文件须是后一个的输入文件。
rule all的input是流程最终的目标文件,从顶部指定目标。fastqc不能和其他的rule串起来,它的输出文件不能作为其它rule的输入文件,所以要单独在rule all里指定。
rule all 指定输出文件{sample_name}.count后,通过顶部目标从上往下确定运行逻辑顺序,首先寻找哪个rule的输出文件是{sample_name}.count,可以看出是rule htseq;那么rule htseq的输入文件{sample_name}_nsorted.bam是哪个rule来的呢,它是rule samtools_sort的输出文件;同理我们可以看出rule samtools_sort的输入文件是从rule hisat2的输出结果;以此类推从顶部倒推的结果是rule all >rule htseq>rule samtools_sort>rule hisat2>rule fastqc,所以运行顺序是与之相反的。
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 (SAMPLES,)= glob_wildcards("{sample}.1_1.fastq.gz") rule all: input: expand('{sample_name}.1_1_fastqc.zip',sample_name=SAMPLES), expand('{sample_name}.count',sample_name=SAMPLES) rule fastqc: input: fq='{sample_name}.1_1.fastq.gz' output: '{sample_name}.1_1_fastqc.zip' log: '{sample_name}.1_1.log' params: outdir='qc' shell: 'fastqc {input[fq]} -o {params[outdir]} 1>{log[0]} 2>&1' rule fastp: input: fq='{sample_name}.1_1.fastq.gz' output: '{sample_name}.1_1_clean.fastq.gz' log: '{sample_name}.fastp.log' params: outdir='fastp' shell: 'fastp -i {input[fq]} -o {params[outdir]}/{output[0]}' rule hisat2: input: clean_fq='{sample_name}.1_1_clean.fastq.gz' output: '{sample_name}.sam' log: '{sample_name}.hisat2.log' params: outdir='hisat2', index='index/hg19/genome' shell: 'hisat2 -x {params[index]} -p 10 -U fastp/{input[clean_fq]} -S {params[outdir]}/{output[0]}' rule samtools_sort: input: '{sample_name}.sam' output: '{sample_name}_nsorted.bam' shell: 'samtools sort -o hisat2/{output[0]} hisat2/{input[0]}' rule htseq: input: '{sample_name}_nsorted.bam' output: '{sample_name}.count' log: '{sample_name}.htseq.log' params: outdir='htseq', gtf='genome/gencode.v19.annotation.gtf' shell: 'htseq-count -f bam -r name hisat2/{input[0]} {params[gtf]} >{output[0]}'
dry run 一下,可以加上 -p 参数让终端打印出 shell 运行的命令:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 144 145 146 147 148 149 150 151 152 153 154 155 156 157 158 159 160 161 162 163 164 165 166 167 168 169 170 171 172 173 174 175 176 177 178 179 180 181 182 183 184 185 186 187 188 189 190 191 192 193 194 195 196 197 198 199 200 201 202 203 204 205 206 207 208 209 210 211 212 213 214 215 216 217 218 219 220 221 222 223 $ snakemake -np -s rnaseqflow.py Building DAG of jobs... Job counts: count jobs 1 all 4 fastp 4 fastqc 4 hisat2 4 htseq 4 samtools_sort 21 [Fri Oct 23 10:23:24 2020] rule fastp: input: SRR957678.1_1.fastq.gz output: SRR957678.1_1_clean.fastq.gz log: SRR957678.fastp.log jobid: 18 wildcards: sample_name=SRR957678 fastp -i SRR957678.1_1.fastq.gz -o fastp/SRR957678.1_1_clean.fastq.gz [Fri Oct 23 10:23:24 2020] rule fastqc: input: SRR957679.1_1.fastq.gz output: SRR957679.1_1_fastqc.zip log: SRR957679.1_1.log jobid: 3 wildcards: sample_name=SRR957679 fastqc SRR957679.1_1.fastq.gz -o qc 1>SRR957679.1_1.log 2>&1 [Fri Oct 23 10:23:24 2020] rule fastqc: input: SRR957680.1_1.fastq.gz output: SRR957680.1_1_fastqc.zip log: SRR957680.1_1.log jobid: 4 wildcards: sample_name=SRR957680 fastqc SRR957680.1_1.fastq.gz -o qc 1>SRR957680.1_1.log 2>&1 [Fri Oct 23 10:23:24 2020] rule fastp: input: SRR957679.1_1.fastq.gz output: SRR957679.1_1_clean.fastq.gz log: SRR957679.fastp.log jobid: 19 wildcards: sample_name=SRR957679 fastp -i SRR957679.1_1.fastq.gz -o fastp/SRR957679.1_1_clean.fastq.gz [Fri Oct 23 10:23:24 2020] rule fastp: input: SRR957677.1_1.fastq.gz output: SRR957677.1_1_clean.fastq.gz log: SRR957677.fastp.log jobid: 17 wildcards: sample_name=SRR957677 fastp -i SRR957677.1_1.fastq.gz -o fastp/SRR957677.1_1_clean.fastq.gz [Fri Oct 23 10:23:24 2020] rule fastqc: input: SRR957678.1_1.fastq.gz output: SRR957678.1_1_fastqc.zip log: SRR957678.1_1.log jobid: 2 wildcards: sample_name=SRR957678 fastqc SRR957678.1_1.fastq.gz -o qc 1>SRR957678.1_1.log 2>&1 [Fri Oct 23 10:23:24 2020] rule fastqc: input: SRR957677.1_1.fastq.gz output: SRR957677.1_1_fastqc.zip log: SRR957677.1_1.log jobid: 1 wildcards: sample_name=SRR957677 fastqc SRR957677.1_1.fastq.gz -o qc 1>SRR957677.1_1.log 2>&1 [Fri Oct 23 10:23:24 2020] rule fastp: input: SRR957680.1_1.fastq.gz output: SRR957680.1_1_clean.fastq.gz log: SRR957680.fastp.log jobid: 20 wildcards: sample_name=SRR957680 fastp -i SRR957680.1_1.fastq.gz -o fastp/SRR957680.1_1_clean.fastq.gz [Fri Oct 23 10:23:24 2020] rule hisat2: input: SRR957679.1_1_clean.fastq.gz output: SRR957679.sam log: SRR957679.hisat2.log jobid: 15 wildcards: sample_name=SRR957679 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957679.1_1_clean.fastq.gz -S hisat2/SRR957679.sam [Fri Oct 23 10:23:24 2020] rule hisat2: input: SRR957680.1_1_clean.fastq.gz output: SRR957680.sam log: SRR957680.hisat2.log jobid: 16 wildcards: sample_name=SRR957680 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957680.1_1_clean.fastq.gz -S hisat2/SRR957680.sam [Fri Oct 23 10:23:24 2020] rule hisat2: input: SRR957678.1_1_clean.fastq.gz output: SRR957678.sam log: SRR957678.hisat2.log jobid: 14 wildcards: sample_name=SRR957678 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957678.1_1_clean.fastq.gz -S hisat2/SRR957678.sam [Fri Oct 23 10:23:24 2020] rule hisat2: input: SRR957677.1_1_clean.fastq.gz output: SRR957677.sam log: SRR957677.hisat2.log jobid: 13 wildcards: sample_name=SRR957677 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957677.1_1_clean.fastq.gz -S hisat2/SRR957677.sam [Fri Oct 23 10:23:24 2020] rule samtools_sort: input: SRR957677.sam output: SRR957677_nsorted.bam jobid: 9 wildcards: sample_name=SRR957677 samtools sort -o hisat2/SRR957677_nsorted.bam hisat2/SRR957677.sam [Fri Oct 23 10:23:24 2020] rule samtools_sort: input: SRR957679.sam output: SRR957679_nsorted.bam jobid: 11 wildcards: sample_name=SRR957679 samtools sort -o hisat2/SRR957679_nsorted.bam hisat2/SRR957679.sam [Fri Oct 23 10:23:24 2020] rule samtools_sort: input: SRR957680.sam output: SRR957680_nsorted.bam jobid: 12 wildcards: sample_name=SRR957680 samtools sort -o hisat2/SRR957680_nsorted.bam hisat2/SRR957680.sam [Fri Oct 23 10:23:24 2020] rule samtools_sort: input: SRR957678.sam output: SRR957678_nsorted.bam jobid: 10 wildcards: sample_name=SRR957678 samtools sort -o hisat2/SRR957678_nsorted.bam hisat2/SRR957678.sam [Fri Oct 23 10:23:24 2020] rule htseq: input: SRR957678_nsorted.bam output: SRR957678.count log: SRR957678.htseq.log jobid: 6 wildcards: sample_name=SRR957678 htseq-count -f bam -r name hisat2/SRR957678_nsorted.bam genome/gencode.v19.annotation.gtf >SRR957678.count [Fri Oct 23 10:23:24 2020] rule htseq: input: SRR957680_nsorted.bam output: SRR957680.count log: SRR957680.htseq.log jobid: 8 wildcards: sample_name=SRR957680 htseq-count -f bam -r name hisat2/SRR957680_nsorted.bam genome/gencode.v19.annotation.gtf >SRR957680.count [Fri Oct 23 10:23:24 2020] rule htseq: input: SRR957677_nsorted.bam output: SRR957677.count log: SRR957677.htseq.log jobid: 5 wildcards: sample_name=SRR957677 htseq-count -f bam -r name hisat2/SRR957677_nsorted.bam genome/gencode.v19.annotation.gtf >SRR957677.count [Fri Oct 23 10:23:24 2020] rule htseq: input: SRR957679_nsorted.bam output: SRR957679.count log: SRR957679.htseq.log jobid: 7 wildcards: sample_name=SRR957679 htseq-count -f bam -r name hisat2/SRR957679_nsorted.bam genome/gencode.v19.annotation.gtf >SRR957679.count [Fri Oct 23 10:23:24 2020] localrule all: input: SRR957677.1_1_fastqc.zip, SRR957678.1_1_fastqc.zip, SRR957679.1_1_fastqc.zip, SRR957680.1_1_fastqc.zip, SRR957677.count, SRR957678.count, SRR957679.count, SRR957680.count jobid: 0 Job counts: count jobs 1 all 4 fastp 4 fastqc 4 hisat2 4 htseq 4 samtools_sort 21 This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
假设我们将rule all 里的{sample_name}.count 改为{sample_name}.sam那么命令只会运行到rule hisat2,跳过之后的命令。顶部倒推,rule all指定目标{sample_name}.sam是rule hisat2的输出文件,所以最后运行的rule是rule hisat2,之后的命令不运行。
1 2 3 4 5 (SAMPLES,)= glob_wildcards("{sample}.1_1.fastq.gz") rule all: input: expand('{sample_name}.1_1_fastqc.zip',sample_name=SAMPLES), expand('{sample_name}.sam',sample_name=SAMPLES)
dry run 一下,可以加上 -p 参数让终端打印出 shell 运行的命令:
1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 16 17 18 19 20 21 22 23 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49 50 51 52 53 54 55 56 57 58 59 60 61 62 63 64 65 66 67 68 69 70 71 72 73 74 75 76 77 78 79 80 81 82 83 84 85 86 87 88 89 90 91 92 93 94 95 96 97 98 99 100 101 102 103 104 105 106 107 108 109 110 111 112 113 114 115 116 117 118 119 120 121 122 123 124 125 126 127 128 129 130 131 132 133 134 135 136 137 138 139 140 141 142 143 $ snakemake -np -s rnaseqflow.py Building DAG of jobs... Job counts: count jobs 1 all 4 fastp 4 fastqc 4 hisat2 13 [Fri Oct 23 10:43:36 2020] rule fastqc: input: SRR957679.1_1.fastq.gz output: SRR957679.1_1_fastqc.zip log: SRR957679.1_1.log jobid: 3 wildcards: sample_name=SRR957679 fastqc SRR957679.1_1.fastq.gz -o qc 1>SRR957679.1_1.log 2>&1 [Fri Oct 23 10:43:36 2020] rule fastqc: input: SRR957680.1_1.fastq.gz output: SRR957680.1_1_fastqc.zip log: SRR957680.1_1.log jobid: 4 wildcards: sample_name=SRR957680 fastqc SRR957680.1_1.fastq.gz -o qc 1>SRR957680.1_1.log 2>&1 [Fri Oct 23 10:43:36 2020] rule fastp: input: SRR957678.1_1.fastq.gz output: SRR957678.1_1_clean.fastq.gz log: SRR957678.fastp.log jobid: 10 wildcards: sample_name=SRR957678 fastp -i SRR957678.1_1.fastq.gz -o fastp/SRR957678.1_1_clean.fastq.gz [Fri Oct 23 10:43:36 2020] rule fastp: input: SRR957680.1_1.fastq.gz output: SRR957680.1_1_clean.fastq.gz log: SRR957680.fastp.log jobid: 12 wildcards: sample_name=SRR957680 fastp -i SRR957680.1_1.fastq.gz -o fastp/SRR957680.1_1_clean.fastq.gz [Fri Oct 23 10:43:36 2020] rule fastqc: input: SRR957678.1_1.fastq.gz output: SRR957678.1_1_fastqc.zip log: SRR957678.1_1.log jobid: 2 wildcards: sample_name=SRR957678 fastqc SRR957678.1_1.fastq.gz -o qc 1>SRR957678.1_1.log 2>&1 [Fri Oct 23 10:43:36 2020] rule fastqc: input: SRR957677.1_1.fastq.gz output: SRR957677.1_1_fastqc.zip log: SRR957677.1_1.log jobid: 1 wildcards: sample_name=SRR957677 fastqc SRR957677.1_1.fastq.gz -o qc 1>SRR957677.1_1.log 2>&1 [Fri Oct 23 10:43:36 2020] rule fastp: input: SRR957677.1_1.fastq.gz output: SRR957677.1_1_clean.fastq.gz log: SRR957677.fastp.log jobid: 9 wildcards: sample_name=SRR957677 fastp -i SRR957677.1_1.fastq.gz -o fastp/SRR957677.1_1_clean.fastq.gz [Fri Oct 23 10:43:36 2020] rule fastp: input: SRR957679.1_1.fastq.gz output: SRR957679.1_1_clean.fastq.gz log: SRR957679.fastp.log jobid: 11 wildcards: sample_name=SRR957679 fastp -i SRR957679.1_1.fastq.gz -o fastp/SRR957679.1_1_clean.fastq.gz [Fri Oct 23 10:43:36 2020] rule hisat2: input: SRR957678.1_1_clean.fastq.gz output: SRR957678.sam log: SRR957678.hisat2.log jobid: 6 wildcards: sample_name=SRR957678 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957678.1_1_clean.fastq.gz -S hisat2/SRR957678.sam [Fri Oct 23 10:43:36 2020] rule hisat2: input: SRR957680.1_1_clean.fastq.gz output: SRR957680.sam log: SRR957680.hisat2.log jobid: 8 wildcards: sample_name=SRR957680 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957680.1_1_clean.fastq.gz -S hisat2/SRR957680.sam [Fri Oct 23 10:43:36 2020] rule hisat2: input: SRR957677.1_1_clean.fastq.gz output: SRR957677.sam log: SRR957677.hisat2.log jobid: 5 wildcards: sample_name=SRR957677 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957677.1_1_clean.fastq.gz -S hisat2/SRR957677.sam [Fri Oct 23 10:43:36 2020] rule hisat2: input: SRR957679.1_1_clean.fastq.gz output: SRR957679.sam log: SRR957679.hisat2.log jobid: 7 wildcards: sample_name=SRR957679 hisat2 -x index/hg19/genome -p 10 -U fastp/SRR957679.1_1_clean.fastq.gz -S hisat2/SRR957679.sam [Fri Oct 23 10:43:36 2020] localrule all: input: SRR957677.1_1_fastqc.zip, SRR957678.1_1_fastqc.zip, SRR957679.1_1_fastqc.zip, SRR957680.1_1_fastqc.zip, SRR957677.sam, SRR957678.sam, SRR957679.sam, SRR957680.sam jobid: 0 Job counts: count jobs 1 all 4 fastp 4 fastqc 4 hisat2 13 This was a dry-run (flag -n). The order of jobs does not reflect the order of execution.
六.生成可视化流程图 Snakemake 可以将整个工作流以流程图的形式导出(结合 dot 命令),文件格式可以使png、pdf等。命令如下:
1 2 $ snakemake --dag -s rnaseqflow.py| dot -Tpdf > test.pdf Building DAG of jobs...
引用 1.Snakemake:简单好用的生信流程搭建工具
2.小白手册:使用Snakemake来轻松搞定生信分析流程