原始数据下载地址:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50177
一.SRA数据下载
打开网页按以下步骤进行
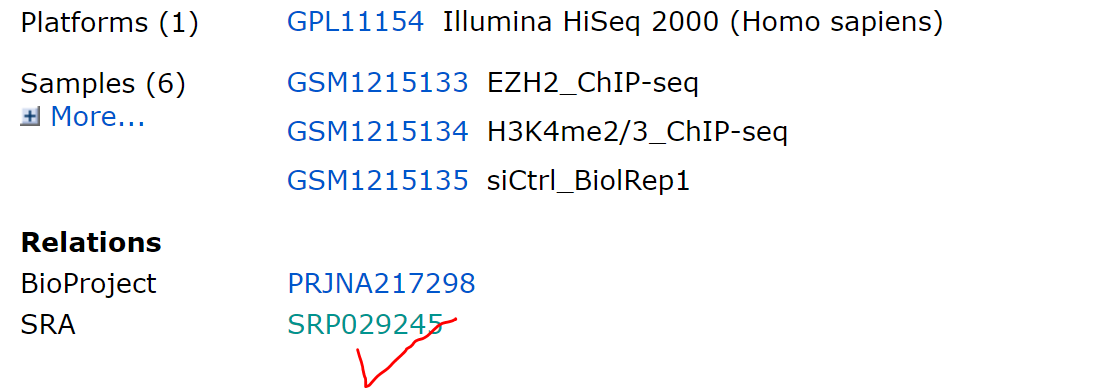
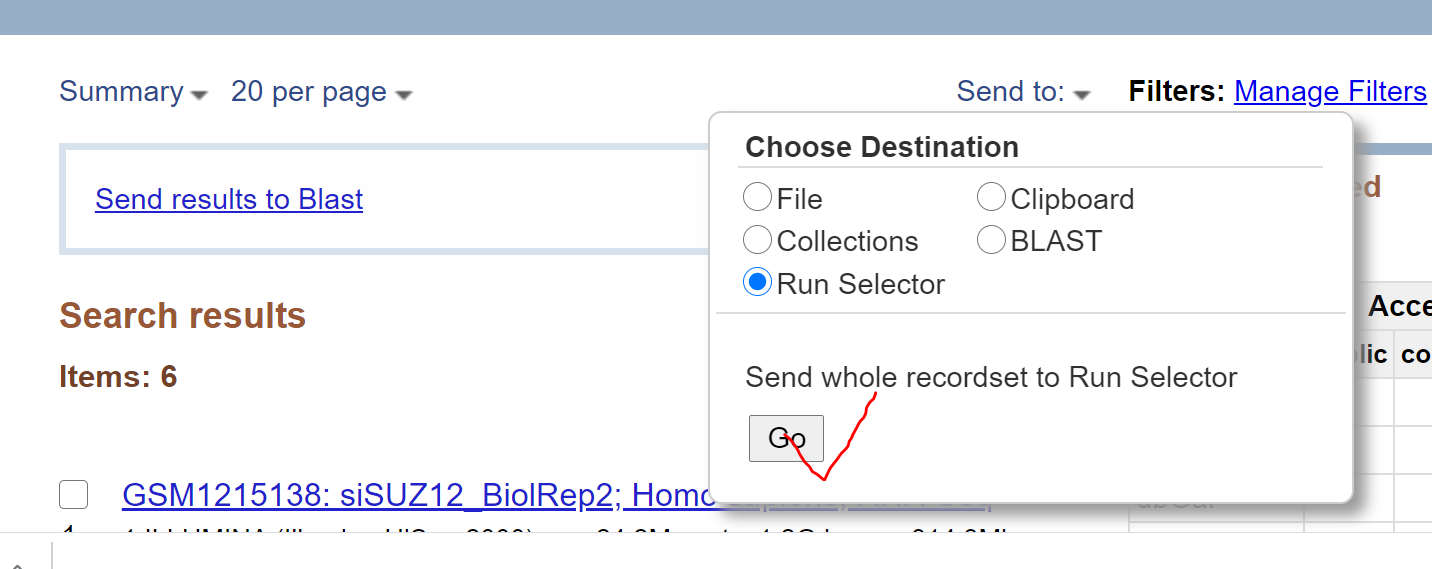
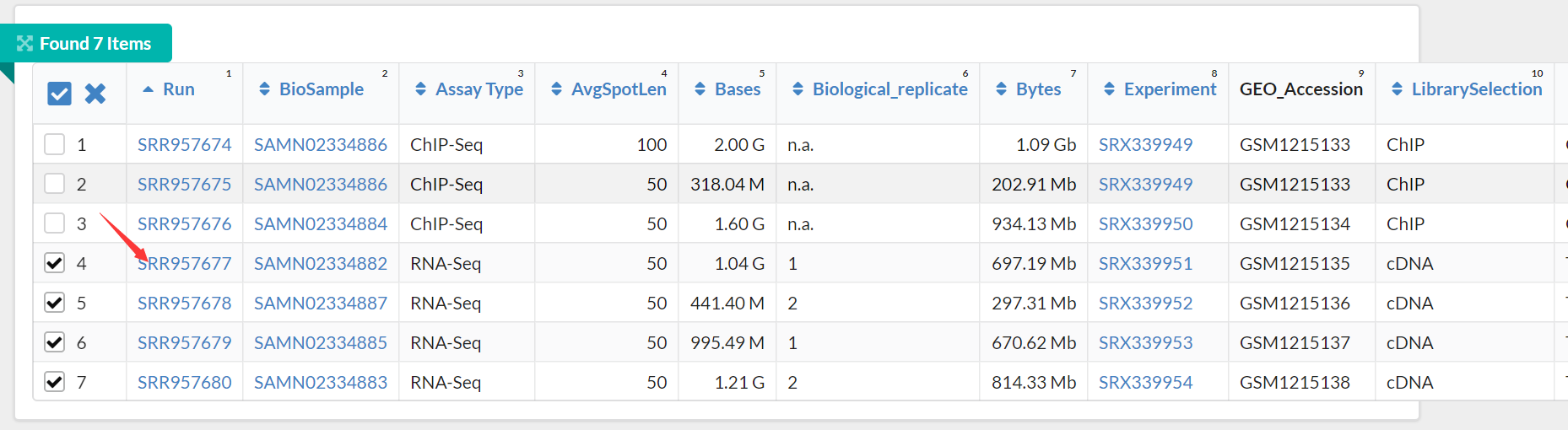
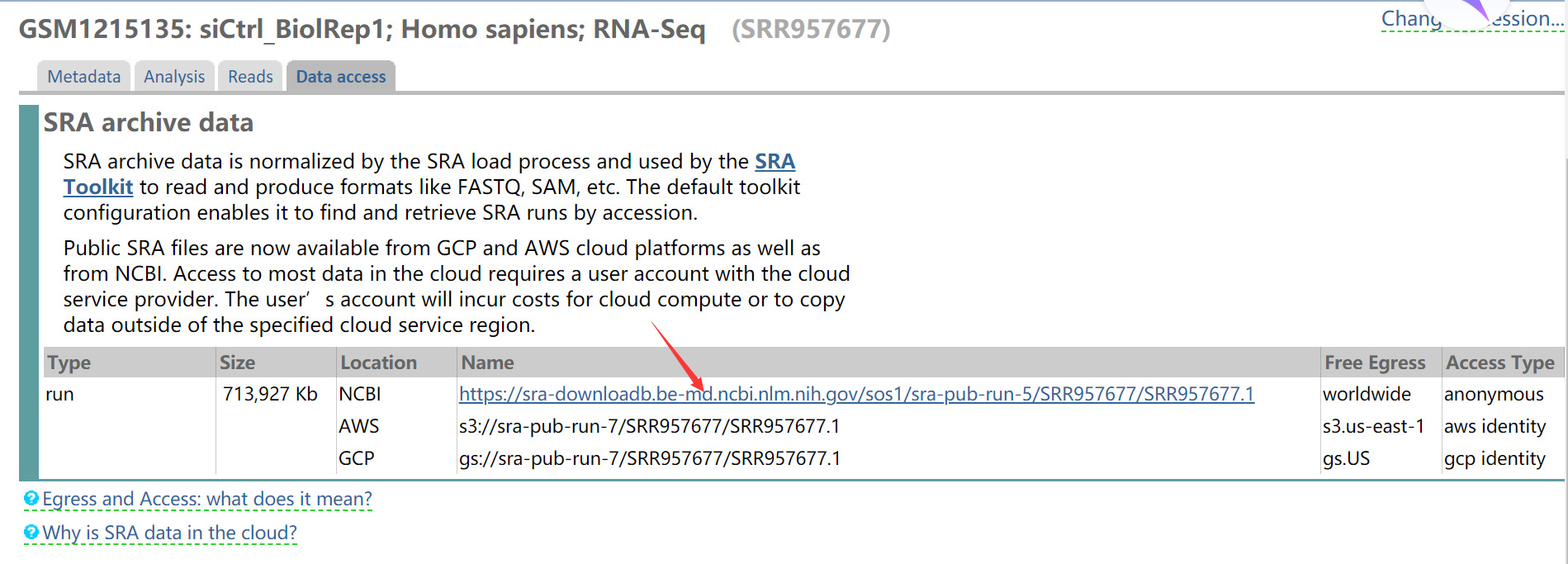
将连接复制到迅雷下载,可以淘宝临时买个会员下载,速度较快,建议使用该方式下载。
也可复制地址用wget下载,速度贼慢,不建议。
1 | wget https://sra-downloadb.be-md.ncbi.nlm.nih.gov/sos1/sra-pub-run-5/SRR957677/SRR957677.1 |
数据下载后通过xftp上传到服务器
二.使用sratoolkit从sra文件提取fastq文件
1 | fastq-dump --gzip --split-files SRR957677 #从SRR957677分别提取左端和右端reads |
–gzip 生成压缩格式为gz的fastq文件
SRR957677是一个PE测序结果,所以,需要–split-files参数可以将其分解为两个fastq文件。如果不加该参数,则只有1个fastq文件(包含了两端测序的结果)
有四个文件分别是SRR957677,SRR957678,SRR957679,SRR957680,;可以一个一个地提取fastq文件,也可以批量提取,有多种方法批量提取。
1)for循环
检查当前文件夹是否有SRR文件,*是匹配任意字符
1 | ls SRR* |
检查循环是否正确,echo打印出脚本
1 | for i in `ls SRR*`;do echo fastq-dump --gzip --split-files $i ;done |
将上述命令中的echo去掉直接运行即可
1 | for i in `ls SRR*`;do fastq-dump --gzip --split-files $i ;done |
2)while循环
新建文件列表这里命名SRR_list
1 | ls SRR* >SRR_list |
查看文件列表是否正确
1 | cat SRR_list |

检查循环是否正确,echo打印出脚本
1 | cat SRR_list | while read id;do echo fastq-dump --gzip --split-files $id ;done |
或者
1 | ls SRR95* | while read id;do echo fastq-dump --gzip --split-files $id ;done#这里改为SRR95*是因为当前有个文件名为SRR_list,如果还是SRR*则会把这个文件也匹配进去 |
3)xargs
1 | ls SRR95* | xargs -i echo echo fastq-dump --gzip --split-files {} |
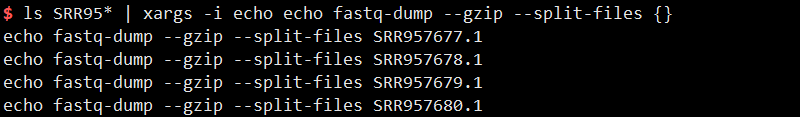
4)awk
1 | awk '{print "nohup fastq-dump --gzip --split-files "$0 " &"}' SRR_list >fastq_dump.sh |
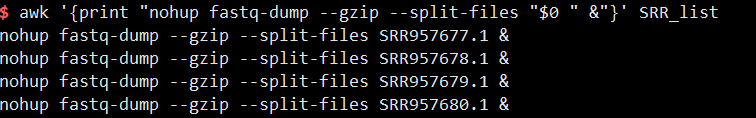
加nohup & 是为了放后台,因为每一个子命令都有nohup &所以这些命令是并行,>fastq_dump.sh是将命令写入fastq_dump.sh中。
1 | bash fastq_dump.sh |
查看命令是否挂后台,top查看进程,-u 参数接用户名
1 | top -u liyue |
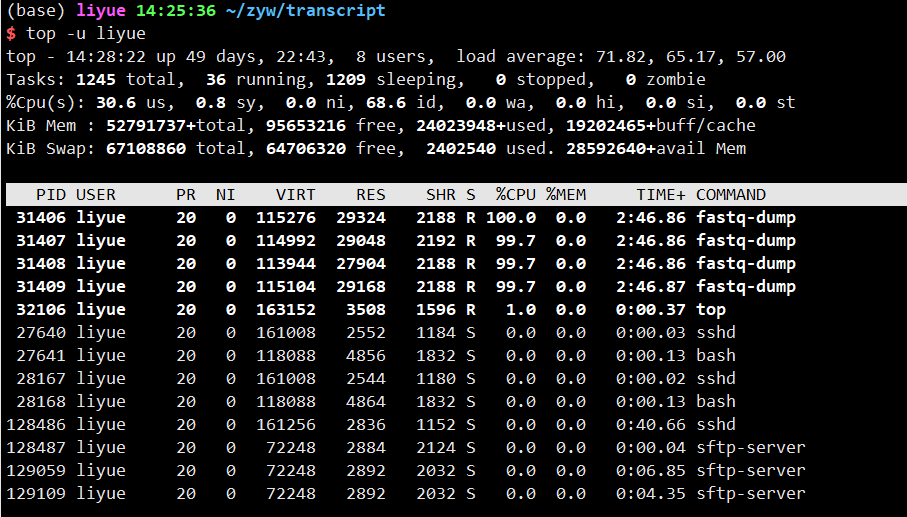
可以看到有四个fastq-dump命令运行中
还可以使用其他命令查询
1 | ps -aux | grep "fastq_dump" |

注意以上命令有个小问题,-split-files参数可以不加,因为这些测序文件是单端测序,所以即使加了该参数也只有一个fastq文件生成,如果是双端测序则会生成左端,右端两个fastq文件,不过加了也没影响
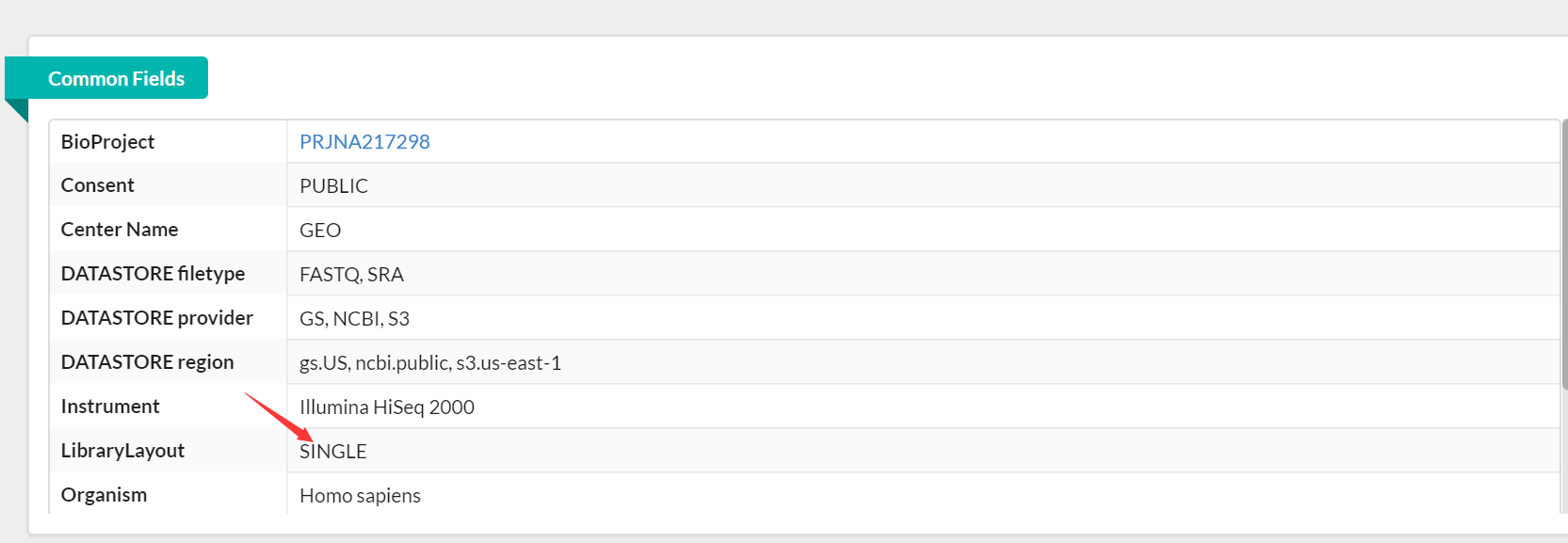
生成以下fastq文件

三.质控
1.质控使用fastqc软件
该软件可直接用conda安装,这里使用循环进行质控
1 | ls *fastq.gz | awk '{print "nohup fastqc " $0 " &"}' >fastqc.sh |
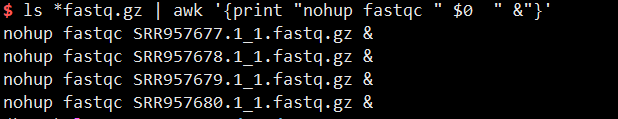
运行fastqc.sh脚本
1 | bash fastqc.sh |
每个fastq文件质控后生一个html和zip压缩格式文件,html可下载到本地用浏览器打开

2.multiqc
可以将当前各个fastqz文件的质控报告综合到一起,生成一个总的html命令multiqc接fastqc结果文件所在地址
1 | multiqc ./ |
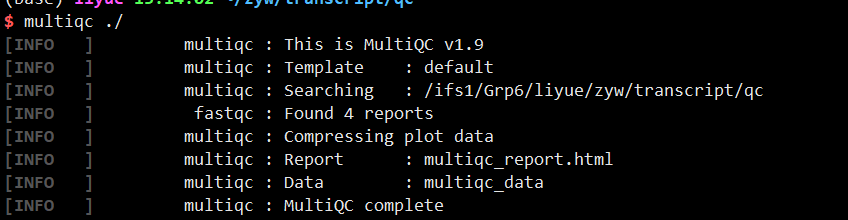
生成结果文件为multiqc_report.html,html可下载到本地用浏览器打开
3.去除低质量碱基
1.fastp
fastp去除低质量碱基的同时可以自动识别接头
单端测序:fastp -i 输入文件 -o 输出文件
双端测序:fastp - i reads1 -I reads2 -o reads1输出文件 -O reads2输出文件
1 | ls *fastq.gz >fastq_list#生成fastq列表 |
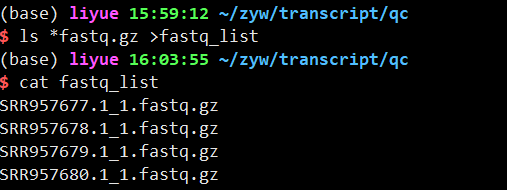

2.trim-galore
1 | trim_galore [options] <filename> |
1 | trim_galore -q 20 --phred33 --stringency 3 --length 20 -e 0.1 \ |
3.fastqc
再次用fastqc检测碱基质量,并与去除低质量碱基前的fastqc结果进行对比

